| PGM3 deficiency | |
|---|---|
| Other names | PGM3-related congenital disorder of glycosylation |
PGM3 deficiency is a rare genetic disorder of the immune system associated with diminished phosphoglucomutase 3 function. PGM3 is an enzyme which in humans is encoded by gene PGM3. This disorder manifests as severe atopy, immune deficiency, autoimmunity, intellectual disability, and hypomyelination. In 2014, Investigators Atfa Sassi at the Pasteur Institute of Tunis, Sandra Lazaroski at the University Medical Center Freiburg, and Gang Wu at the Imperial College London, identified PGM3 mutations in nine patients from four consanguineous families.[1] In the same year, a researchers from the laboratories of Joshua Milner [2] and Helen Su [3] at the National Institute of Allergy and Infectious Disease at the U.S. National Institutes of Health described PGM3 deficiency in eight additional patients from two families.[4]
Signs and symptoms
Clinically, PGM3 deficient patients are marked with a group of immunologic and neurologic impairments. Often patients present with similar manifestations to Hyperimmunoglobulin E syndrome (HIES), including severe atopic dermatitis, chronic sinusitis or otitis, cutaneous vasculitis, severe pulmonary infections and pneumonia, and very high concentrations of the serum antibody IgE levels. Skin infections are prominent, with recurrent staphylococcal and fungal infections, severe skin blistering, and flat warts. Patients are susceptible to reoccurring pulmonary infections that may lead to bronchiectasis in the future. Additionally, scoliosis and microcephaly have also been identified.[1][4][5]
In addition to severe immunodeficiency, motor and neurologic impairment are evident from early life. Oral motor deficits, dysarthria, developmental delay, ataxia, myoclonus, seizure and mild sensory loss have all been identified. These distinctive neurologic features are suggestive of hypomyelination, as they resemble features of other congenital disorder of glycosylation (CDGs). Because glycosylation is known to be critical for numerous immune-related proteins, these patients likely present with additional abnormalities including hemolytic anemia, hepatosplenomegaly, and neutropenia. An immunologic mechanism to explain the link between glycosylation abnormalities and the immune dysregulation has not yet been established. This disorder demonstrates a previously unappreciated importance that glycosylation can have on the immune response and more research is needed to examine the precise mechanism by which these mutations and abnormal glycosylation lead to the clinical defects observed [1][4][5]
Genetics
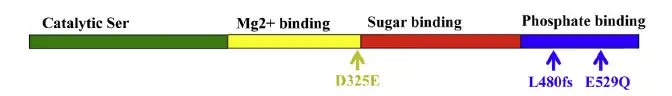
PGM3 deficiency is caused by a hypomorphic mutation in gene PGM3 (OMIM#172100). PGM3 is a 29 kb gene with 14 exons, mapping to chromosome 6q14.1-q14.2 and encoding a 542 amino acid protein that serves as the crucial catalyst for the glycosylation pathway [6][7] Protein PGM3 is required for the reversible conversion of GlcNAc-6-phosphate (GlnNAc-6-P) to GlnNAc-a-phosphate (GlcNac-1-P), a precursor step for the synthesis of uridine diphosphate N-acetylglucosamine (UDP-GlcNAc). Impaired function of PGM3 is demonstrated by decreased enzyme activity, reduced UDP-GlcNAc and reduced N-linked glycosylation and O-linked glycosylation[4]
PGM3 is composed of four protein domains: active serine domain, metal-binding domain, sugar-binding domain, and phosphate-binding domain. Missense and deletion mutations have been discovered within multiple of these domains. All identified mutant PGM3 retain catalytic specificity however weaken enzymatic activity. Mutations in the sugar-binding domain leads to reduced PGM3 abundance and impairs PGM3 function and glycosylation to a higher extent than mutations in the catalytic or phosphate-binding domain, and results in a more severe clinical phenotype.[1]
Inheritance

PGM3 deficiency is inherited in an autosomal recessive manner. Autosomal refers to the fact that every person has two PGM3 alleles, one inherited from each parent. Recessive refers to each affected person needs two copies of the abnormal gene—one copy from each parent—to develop the syndrome.
Typically, both parents of an affected child carry one abnormal gene and are unaffected by the disease. When both parents have one abnormal copy of the PGM3 gene, each child has a 25 percent chance of being affected by the disease.
Sometimes the two copies of the PGM3 gene that a child inherits have identical, or homozygous, mutations. This is common if the child’s parents are related to each other, explaining why many reported cases of PGM3 deficiency have involved consanguinity families. Many patients have different mutations on the two copies of PGM3, and their mutations are called compound heterozygous mutations. In either case, the patient is not able to produce functional PGM3 protein
Diagnosis
Laboratory manifestations
Patients have significantly increased levels of serum IgE, IgG, and IgA. Additionally, patients have significant leukopenia, lymphopenia, neutropenia, and eosinophilia. Mild defects in T-cell function can also be observed, in addition to an inverted CD4/CD8 ratio [4]
Treatment
Once a diagnosis is made, the treatment is based on an individual’s clinical condition and may include standard management for autoimmunity and immunodeficiency. It has been suggested that exogenous nondiabeotgenic sugar supplementations of GlcNAc might be used to increase UDP-GlcNAc and bypass the metabolic defect for treatment.[4] Investigators at the National Institute of Allergy and Infectious Diseases at the US National Institutes of Health currently have clinical protocols to study new approaches to the diagnosis and treatment of this disorder.[8]
References
- 1 2 3 4 Sassi A, Lazaroski S, Wu G, Haslam SM, Fliegauf M, Mellouli F, et al. (May 2014). "Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels". The Journal of Allergy and Clinical Immunology. 133 (5): 1410–9, 1419.e1–13. doi:10.1016/j.jaci.2014.02.025. PMC 4825677. PMID 24698316.
- ↑ "Joshua Milner, M.D." NIH.
- ↑ "Helen Su, M.D". NIH.
- 1 2 3 4 5 6 7 Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, et al. (May 2014). "Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment". The Journal of Allergy and Clinical Immunology. 133 (5): 1400–9, 1409.e1–5. doi:10.1016/j.jaci.2014.02.013. PMC 4016982. PMID 24589341.
- 1 2 Hay BN, Martin JE, Karp B, Davis J, Darnell D, Solomon B, Turner M, Holland SM, Puck JM (March 2004). "Familial immunodeficiency with cutaneous vasculitis, myoclonus, and cognitive impairment". American Journal of Medical Genetics. Part A. 125A (2): 145–51. doi:10.1002/ajmg.a.20595. PMID 14981714. S2CID 34548634.
- ↑ "OMIN Entry#172100-PHOSPHOGLUCOMUTASE 3; PGM3".
- ↑ "PGM3 Gene". GeneCards.
- ↑ "study ID#NCT02511041". ClinicalTrials.gov. 30 June 2017.