Chromatin remodeling is the dynamic modification of chromatin architecture to allow access of condensed genomic DNA to the regulatory transcription machinery proteins, and thereby control gene expression. Such remodeling is principally carried out by 1) covalent histone modifications by specific enzymes, e.g., histone acetyltransferases (HATs), deacetylases, methyltransferases, and kinases, and 2) ATP-dependent chromatin remodeling complexes which either move, eject or restructure nucleosomes.[1] Besides actively regulating gene expression, dynamic remodeling of chromatin imparts an epigenetic regulatory role in several key biological processes, egg cells DNA replication and repair; apoptosis; chromosome segregation as well as development and pluripotency. Aberrations in chromatin remodeling proteins are found to be associated with human diseases, including cancer. Targeting chromatin remodeling pathways is currently evolving as a major therapeutic strategy in the treatment of several cancers.
Overview
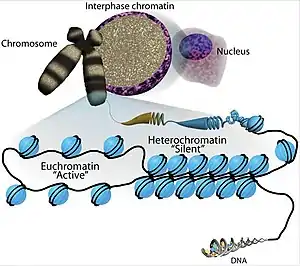
The transcriptional regulation of the genome is controlled primarily at the preinitiation stage by binding of the core transcriptional machinery proteins (namely, RNA polymerase, transcription factors, and activators and repressors) to the core promoter sequence on the coding region of the DNA. However, DNA is tightly packaged in the nucleus with the help of packaging proteins, chiefly histone proteins to form repeating units of nucleosomes which further bundle together to form condensed chromatin structure. Such condensed structure occludes many DNA regulatory regions, not allowing them to interact with transcriptional machinery proteins and regulate gene expression. To overcome this issue and allow dynamic access to condensed DNA, a process known as chromatin remodeling alters nucleosome architecture to expose or hide regions of DNA for transcriptional regulation.
By definition, chromatin remodeling is the enzyme-assisted process to facilitate access of nucleosomal DNA by remodeling the structure, composition and positioning of nucleosomes.
Classification
Access to nucleosomal DNA is governed by two major classes of protein complexes:
- Covalent histone-modifying complexes.
- ATP-dependent chromatin remodeling complexes.
Covalent histone-modifying complexes
Specific protein complexes, known as histone-modifying complexes catalyze addition or removal of various chemical elements on histones. These enzymatic modifications include acetylation, methylation, phosphorylation, and ubiquitination and primarily occur at N-terminal histone tails. Such modifications affect the binding affinity between histones and DNA, and thus loosening or tightening the condensed DNA wrapped around histones, e.g., Methylation of specific lysine residues in H3 and H4 causes further condensation of DNA around histones, and thereby prevents binding of transcription factors to the DNA that lead to gene repression. On the contrary, histone acetylation relaxes chromatin condensation and exposes DNA for TF binding, leading to increased gene expression.[3]
Known modifications
Well characterized modifications to histones include:[4]
Both lysine and arginine residues are known to be methylated. Methylated lysines are the best understood marks of the histone code, as specific methylated lysine match well with gene expression states. Methylation of lysines H3K4 and H3K36 is correlated with transcriptional activation while demethylation of H3K4 is correlated with silencing of the genomic region. Methylation of lysines H3K9 and H3K27 is correlated with transcriptional repression.[5] Particularly, H3K9me3 is highly correlated with constitutive heterochromatin.[6]
- Acetylation - by HAT (histone acetyl transferase); deacetylation - by HDAC (histone deacetylase)
Acetylation tends to define the 'openness' of chromatin as acetylated histones cannot pack as well together as deacetylated histones.
However, there are many more histone modifications, and sensitive mass spectrometry approaches have recently greatly expanded the catalog.[7]
Histone code hypothesis
The histone code is a hypothesis that the transcription of genetic information encoded in DNA is in part regulated by chemical modifications to histone proteins, primarily on their unstructured ends. Together with similar modifications such as DNA methylation it is part of the epigenetic code.
Cumulative evidence suggests that such code is written by specific enzymes which can (for example) methylate or acetylate DNA ('writers'), removed by other enzymes having demethylase or deacetylase activity ('erasers'), and finally readily identified by proteins ('readers') that are recruited to such histone modifications and bind via specific domains, e.g., bromodomain, chromodomain. These triple action of 'writing', 'reading' and 'erasing' establish the favorable local environment for transcriptional regulation, DNA-damage repair, etc.[8]
The critical concept of the histone code hypothesis is that the histone modifications serve to recruit other proteins by specific recognition of the modified histone via protein domains specialized for such purposes, rather than through simply stabilizing or destabilizing the interaction between histone and the underlying DNA. These recruited proteins then act to alter chromatin structure actively or to promote transcription.
A very basic summary of the histone code for gene expression status is given below (histone nomenclature is described here):
| Type of modification |
Histone | ||||||
|---|---|---|---|---|---|---|---|
| H3K4 | H3K9 | H3K14 | H3K27 | H3K79 | H4K20 | H2BK5 | |
| mono-methylation | activation[9] | activation[10] | activation[10] | activation[10][11] | activation[10] | activation[10] | |
| di-methylation | repression[5] | repression[5] | activation[11] | ||||
| tri-methylation | activation[12] | repression[10] | repression[10] | activation,[11] repression[10] |
repression[5] | ||
| acetylation | activation[12] | activation[12] | |||||
ATP-dependent chromatin remodeling
ATP-dependent chromatin-remodeling complexes regulate gene expression by either moving, ejecting or restructuring nucleosomes. These protein complexes have a common ATPase domain and energy from the hydrolysis of ATP allows these remodeling complexes to reposition nucleosomes (often referred to as "nucleosome sliding") along the DNA, eject or assemble histones on/off of DNA or facilitate exchange of histone variants, and thus creating nucleosome-free regions of DNA for gene activation.[13] Also, several remodelers have DNA-translocation activity to carry out specific remodeling tasks.[14]
All ATP-dependent chromatin-remodeling complexes possess a sub unit of ATPase that belongs to the SNF2 superfamily of proteins. In association to the sub unit's identity, two main groups have been classified for these proteins. These are known as the SWI2/SNF2 group and the imitation SWI (ISWI) group. The third class of ATP-dependent complexes that has been recently described contains a Snf2-like ATPase and also demonstrates deacetylase activity.[15]
Known chromatin remodeling complexes
There are at least four families of chromatin remodelers in eukaryotes: SWI/SNF, ISWI, NuRD/Mi-2/CHD, and INO80 with first two remodelers being very well studied so far, especially in the yeast model. Although all of remodelers share common ATPase domain, their functions are specific based on several biological processes (DNA repair, apoptosis, etc.). This is due to the fact that each remodeler complex has unique protein domains (Helicase, bromodomain, etc.) in their catalytic ATPase region and also has different recruited subunits.
Specific functions
- Several in-vitro experiments suggest that ISWI remodelers organize nucleosome into proper bundle form and create equal spacing between nucleosomes, whereas SWI/SNF remodelers disorder nucleosomes.
- The ISWI-family remodelers have been shown to play central roles in chromatin assembly after DNA replication and maintenance of higher-order chromatin structures.
- INO80 and SWI/SNF-family remodelers participate in DNA double-strand break (DSB) repair and nucleotide-excision repair (NER) and thereby plays crucial role in TP53 mediated DNA-damage response.
- NuRD/Mi-2/CHD remodeling complexes primarily mediate transcriptional repression in the nucleus and are required for the maintenance of pluripotency of embryonic stem cells.[13]
Significance
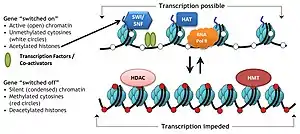
In normal biological processes
Chromatin remodeling plays a central role in the regulation of gene expression by providing the transcription machinery with dynamic access to an otherwise tightly packaged genome. Further, nucleosome movement by chromatin remodelers is essential to several important biological processes, including chromosome assembly and segregation, DNA replication and repair, embryonic development and pluripotency, and cell-cycle progression. Deregulation of chromatin remodeling causes loss of transcriptional regulation at these critical check-points required for proper cellular functions, and thus causes various disease syndromes, including cancer.
Response to DNA damage
Chromatin relaxation is one of the earliest cellular responses to DNA damage.[16] Several experiments have been performed on the recruitment kinetics of proteins involved in the response to DNA damage. The relaxation appears to be initiated by PARP1, whose accumulation at DNA damage is half complete by 1.6 seconds after DNA damage occurs.[17] This is quickly followed by accumulation of chromatin remodeler Alc1, which has an ADP-ribose–binding domain, allowing it to be quickly attracted to the product of PARP1. The maximum recruitment of Alc1 occurs within 10 seconds of DNA damage.[16] About half of the maximum chromatin relaxation, presumably due to action of Alc1, occurs by 10 seconds.[16] PARP1 action at the site of a double-strand break allows recruitment of the two DNA repair enzymes MRE11 and NBS1. Half maximum recruitment of these two DNA repair enzymes takes 13 seconds for MRE11 and 28 seconds for NBS1.[17]
Another process of chromatin relaxation, after formation of a DNA double-strand break, employs γH2AX, the phosphorylated form of the H2AX protein. The histone variant H2AX constitutes about 10% of the H2A histones in human chromatin.[18] γH2AX (phosphorylated on serine 139 of H2AX) was detected at 20 seconds after irradiation of cells (with DNA double-strand break formation), and half maximum accumulation of γH2AX occurred in one minute.[18] The extent of chromatin with phosphorylated γH2AX is about two million base pairs at the site of a DNA double-strand break.[18]
γH2AX does not, by itself, cause chromatin decondensation, but within seconds of irradiation the protein "Mediator of the DNA damage checkpoint 1" (MDC1) specifically attaches to γH2AX.[19][20] This is accompanied by simultaneous accumulation of RNF8 protein and the DNA repair protein NBS1 which bind to MDC1 as MDC1 attaches to γH2AX.[21] RNF8 mediates extensive chromatin decondensation, through its subsequent interaction with CHD4 protein,[22] a component of the nucleosome remodeling and deacetylase complex NuRD. CHD4 accumulation at the site of the double-strand break is rapid, with half-maximum accumulation occurring by 40 seconds after irradiation.[23]
The fast initial chromatin relaxation upon DNA damage (with rapid initiation of DNA repair) is followed by a slow recondensation, with chromatin recovering a compaction state close to its pre-damage level in ~ 20 min.[16]
Cancer
Chromatin remodeling provides fine-tuning at crucial cell growth and division steps, like cell-cycle progression, DNA repair and chromosome segregation, and therefore exerts tumor-suppressor function. Mutations in such chromatin remodelers and deregulated covalent histone modifications potentially favor self-sufficiency in cell growth and escape from growth-regulatory cell signals - two important hallmarks of cancer.[24]
- Inactivating mutations in SMARCB1, formerly known as hSNF5/INI1 and a component of the human SWI/SNF remodeling complex have been found in large number of rhabdoid tumors, commonly affecting pediatric population.[25] Similar mutations are also present in other childhood cancers, such as choroid plexus carcinoma, medulloblastoma and in some acute leukemias. Further, mouse knock-out studies strongly support SMARCB1 as a tumor suppressor protein. Since the original observation of SMARCB1 mutations in rhabdoid tumors, several more subunits of the human SWI/SNF chromatin remodeling complex have been found mutated in a wide range of neoplasms.[26]
- The SWI/SNF ATPase BRG1 (or SMARCA4) is the most frequently mutated chromatin remodeling ATPase in cancer.[27] Mutations in this gene were first recognized in human cancer cell lines derived from lung.[28] In cancer, mutations in BRG1 show an unusually high preference for missense mutations that target the ATPase domain.[29][27] Mutations are enriched at highly conserved ATPase sequences,[30] which lie on important functional surfaces such as the ATP pocket or DNA-binding surface.[29] These mutations act in a genetically dominant manner to alter chromatin regulatory function at enhancers[29] and promoters.[30]
- Inactivating mutations in BCL7A in Diffuse large B-cell lymphoma (DLBCL) [31] and in other haematological malignancies [32]
- PML-RARA fusion protein in acute myeloid leukemia recruits histone deacetylases. This leads to repression of genes responsible for myelocytes to differentiate, leading to leukemia.[33]
- Tumor suppressor Rb protein functions by the recruitment of the human homologs of the SWI/SNF enzymes BRG1, histone deacetylase and DNA methyltransferase. Mutations in BRG1 are reported in several cancers causing loss of tumor suppressor action of Rb.[34]
- Recent reports indicate DNA hypermethylation in the promoter region of major tumor suppressor genes in several cancers. Although few mutations are reported in histone methyltransferases yet, correlation of DNA hypermethylation and histone H3 lysine-9 methylation has been reported in several cancers, mainly in colorectal and breast cancers.
- Mutations in Histone Acetyl Transferases (HAT) p300 (missense and truncating type) are most commonly reported in colorectal, pancreatic, breast and gastric carcinomas. Loss of heterozygosity in coding region of p300 (chromosome 22q13) is present in large number of glioblastomas.
- Further, HATs have diverse role as transcription factors beside having histone acetylase activity, e.g., HAT subunit, hADA3 may act as an adaptor protein linking transcription factors with other HAT complexes. In the absence of hADA3, TP53 transcriptional activity is significantly reduced, suggesting role of hADA3 in activating TP53 function in response to DNA damage.
- Similarly, TRRAP, the human homolog to yeast Tra1, has been shown to directly interact with c-Myc and E2F1, known oncoproteins.[35]
Cancer genomics
Rapid advance in cancer genomics and high-throughput ChIP-chip, ChIP-Seq and Bisulfite sequencing methods are providing more insight into role of chromatin remodeling in transcriptional regulation and role in cancer.
Therapeutic intervention
Epigenetic instability caused by deregulation in chromatin remodeling is studied in several cancers, including breast cancer, colorectal cancer, pancreatic cancer. Such instability largely cause widespread silencing of genes with primary impact on tumor-suppressor genes. Hence, strategies are now being tried to overcome epigenetic silencing with synergistic combination of HDAC inhibitors or HDI and DNA-demethylating agents. HDIs are primarily used as adjunct therapy in several cancer types.[36][37] HDAC inhibitors can induce p21 (WAF1) expression, a regulator of p53's tumor suppressoractivity. HDACs are involved in the pathway by which the retinoblastoma protein (pRb) suppresses cell proliferation.[38] Estrogen is well-established as a mitogenic factor implicated in the tumorigenesis and progression of breast cancer via its binding to the estrogen receptor alpha (ERα). Recent data indicate that chromatin inactivation mediated by HDAC and DNA methylation is a critical component of ERα silencing in human breast cancer cells.[39]
- Approved usage:
- Vorinostat was licensed by the U.S. FDA in October 2006 for the treatment of cutaneous T cell lymphoma (CTCL).[40][41]
- Romidepsin (trade name Istodax) was licensed by the US FDA in Nov 2009 for cutaneous T-cell lymphoma (CTCL).[42][43]
- Phase III Clinical trials:
- Panobinostat (LBH589) is in clinical trials for various cancers including a phase III trial for cutaneous T cell lymphoma (CTCL).
- Valproic acid (as Mg valproate) in phase III trials for cervical cancer and ovarian cancer.
- Started pivotal phase II clinical trials:
- Belinostat (PXD101) has had a phase II trial for relapsed ovarian cancer, and reported good results for T cell lymphoma.
- HDAC inhibitors.
Current front-runner candidates for new drug targets are Histone Lysine Methyltransferases (KMT) and Protein Arginine Methyltransferases (PRMT).[44]
Other disease syndromes
- ATRX-syndrome (α-thalassemia X-linked mental retardation) and α-thalassemia myelodysplasia syndrome are caused by mutations in ATRX, a SNF2-related ATPase with a PHD finger domain.[45]
- CHARGE syndrome, an autosomal dominant disorder, has been linked recently to haploinsufficiency of CHD7, which encodes the CHD family ATPase CHD7.[46]
Senescence
Chromatin architectural remodeling is implicated in the process of cellular senescence, which is related to, and yet distinct from, organismal aging. Replicative cellular senescence refers to a permanent cell cycle arrest where post-mitotic cells continue to exist as metabolically active cells but fail to proliferate.[47][48] Senescence can arise due to age associated degradation, telomere attrition, progerias, pre-malignancies, and other forms of damage or disease. Senescent cells undergo distinct repressive phenotypic changes, potentially to prevent the proliferation of damaged or cancerous cells, with modified chromatin organization, fluctuations in remodeler abundance, and changes in epigenetic modifications.[49][50][47] Senescent cells undergo chromatin landscape modifications as constitutive heterochromatin migrates to the center of the nucleus and displaces euchromatin and facultative heterochromatin to regions at the edge of the nucleus. This disrupts chromatin-lamin interactions and inverts of the pattern typically seen in a mitotically active cell.[51][49] Individual Lamin-Associated Domains (LADs) and Topologically Associating Domains (TADs) are disrupted by this migration which can affect cis interactions across the genome.[52] Additionally, there is a general pattern of canonical histone loss, particularly in terms of the nucleosome histones H3 and H4 and the linker histone H1.[51] Histone variants with two exons are upregulated in senescent cells to produce modified nucleosome assembly which contributes to chromatin permissiveness to senescent changes.[52] Although transcription of variant histone proteins may be elevated, canonical histone proteins are not expressed as they are only made during the S phase of the cell cycle and senescent cells are post-mitotic.[51] During senescence, portions of chromosomes can be exported from the nucleus for lysosomal degradation which results in greater organizational disarray and disruption of chromatin interactions.[50]
Chromatin remodeler abundance may be implicated in cellular senescence as knockdown or knockout of ATP-dependent remodelers such as NuRD, ACF1, and SWI/SNP can result in DNA damage and senescent phenotypes in yeast, C. elegans, mice, and human cell cultures.[53][50][54] ACF1 and NuRD are downregulated in senescent cells which suggests that chromatin remodeling is essential for maintaining a mitotic phenotype.[53][54] Genes involved in signaling for senescence can be silenced by chromatin confirmation and polycomb repressive complexes as seen in PRC1/PCR2 silencing of p16.[55][56] Specific remodeler depletion results in activation of proliferative genes through a failure to maintain silencing.[50] Some remodelers act on enhancer regions of genes rather than the specific loci to prevent re-entry into the cell cycle by forming regions of dense heterochromatin around regulatory regions.[56]
Senescent cells undergo widespread fluctuations in epigenetic modifications in specific chromatin regions compared to mitotic cells. Human and murine cells undergoing replicative senescence experience a general global decrease in methylation; however, specific loci can differ from the general trend.[57][52][50][55] Specific chromatin regions, especially those around the promoters or enhancers of proliferative loci, may exhibit elevated methylation states with an overall imbalance of repressive and activating histone modifications.[49] Proliferative genes may show increases in the repressive mark H3K27me3 while genes involved in silencing or aberrant histone products may be enriched with the activating modification H3K4me3.[52] Additionally, upregulating histone deacetylases, such as members of the sirtuin family, can delay senescence by removing acetyl groups that contribute to greater chromatin accessibility.[58] General loss of methylation, combined with the addition of acetyl groups results in a more accessible chromatin conformation with a propensity towards disorganization when compared to mitotically active cells.[50] General loss of histones precludes addition of histone modifications and contributes changes in enrichment in some chromatin regions during senescence.[51]
See also
- Epigenetics
- Histone
- Nucleosomes
- Chromatin
- Histone acetyltransferase
- Transcription factors
- CAF-1 (Chromatin assembly factor-1) - histone chaperone that execute a coordinating role in сhromatin remodeling.
References
- ↑ Teif VB, Rippe K (September 2009). "Predicting nucleosome positions on the DNA: combining intrinsic sequence preferences and remodeler activities". Nucleic Acids Research. 37 (17): 5641–55. doi:10.1093/nar/gkp610. PMC 2761276. PMID 19625488.
- ↑ Boyer (2009). "The chromatin signature of pluripotent cells". Stembook. doi:10.3824/stembook.1.45.1. PMID 20614601.
- ↑ Wang GG, Allis CD, Chi P (September 2007). "Chromatin remodeling and cancer, Part I: Covalent histone modifications". Trends in Molecular Medicine. 13 (9): 363–72. doi:10.1016/j.molmed.2007.07.003. PMID 17822958.
- ↑ Strahl BD, Allis CD (January 2000). "The language of covalent histone modifications". Nature. 403 (6765): 41–5. Bibcode:2000Natur.403...41S. doi:10.1038/47412. PMID 10638745. S2CID 4418993.
- 1 2 3 4 Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ (March 2009). "Determination of enriched histone modifications in non-genic portions of the human genome". BMC Genomics. 10: 143. doi:10.1186/1471-2164-10-143. PMC 2667539. PMID 19335899.
- ↑ Hublitz P, Albert M, Peters A (28 April 2009). "Mechanisms of Transcriptional Repression by Histone Lysine Methylation". The International Journal of Developmental Biology. 10 (1387): 335–354. doi:10.1387/ijdb.082717ph. ISSN 1696-3547. PMID 19412890.
- ↑ Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y (September 2011). "Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification". Cell. 146 (6): 1016–28. doi:10.1016/j.cell.2011.08.008. PMC 3176443. PMID 21925322.
- ↑ Jenuwein T, Allis CD (August 2001). "Translating the histone code". Science. 293 (5532): 1074–80. CiteSeerX 10.1.1.453.900. doi:10.1126/science.1063127. PMID 11498575. S2CID 1883924.
- ↑ Benevolenskaya EV (August 2007). "Histone H3K4 demethylases are essential in development and differentiation". Biochemistry and Cell Biology. 85 (4): 435–43. doi:10.1139/o07-057. PMID 17713579.
- 1 2 3 4 5 6 7 8 Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K (May 2007). "High-resolution profiling of histone methylations in the human genome". Cell. 129 (4): 823–37. doi:10.1016/j.cell.2007.05.009. PMID 17512414. S2CID 6326093.
- 1 2 3 Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, Blobel GA, Vakoc CR (April 2008). "DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells". Molecular and Cellular Biology. 28 (8): 2825–39. doi:10.1128/MCB.02076-07. PMC 2293113. PMID 18285465.
- 1 2 3 Koch CM, Andrews RM, Flicek P, Dillon SC, Karaöz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P, James KD, Lefebvre GC, Bruce AW, Dovey OM, Ellis PD, Dhami P, Langford CF, Weng Z, Birney E, Carter NP, Vetrie D, Dunham I (June 2007). "The landscape of histone modifications across 1% of the human genome in five human cell lines". Genome Research. 17 (6): 691–707. doi:10.1101/gr.5704207. PMC 1891331. PMID 17567990.
- 1 2 Wang GG, Allis CD, Chi P (September 2007). "Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling". Trends in Molecular Medicine. 13 (9): 373–80. doi:10.1016/j.molmed.2007.07.004. PMC 4337864. PMID 17822959.
- ↑ Saha A, Wittmeyer J, Cairns BR (June 2006). "Chromatin remodelling: the industrial revolution of DNA around histones". Nature Reviews Molecular Cell Biology. 7 (6): 437–47. doi:10.1038/nrm1945. PMID 16723979. S2CID 6180120.
- ↑ Vignali, M.; Hassan, A. H.; Neely, K. E.; Workman, J. L. (2000-03-15). "ATP-Dependent Chromatin-Remodeling Complexes". Molecular and Cellular Biology. 20 (6): 1899–1910. doi:10.1128/mcb.20.6.1899-1910.2000. ISSN 0270-7306. PMC 110808. PMID 10688638.
- 1 2 3 4 Sellou H, Lebeaupin T, Chapuis C, Smith R, Hegele A, Singh HR, Kozlowski M, Bultmann S, Ladurner AG, Timinszky G, Huet S (2016). "The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage". Mol. Biol. Cell. 27 (24): 3791–3799. doi:10.1091/mbc.E16-05-0269. PMC 5170603. PMID 27733626.
- 1 2 Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG (2008). "PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites". J. Biol. Chem. 283 (2): 1197–208. doi:10.1074/jbc.M706734200. PMID 18025084.
- 1 2 3 Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998). "DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139". J. Biol. Chem. 273 (10): 5858–68. doi:10.1074/jbc.273.10.5858. PMID 9488723.
- ↑ Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J (2007). "RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins". Cell. 131 (5): 887–900. doi:10.1016/j.cell.2007.09.040. PMID 18001824. S2CID 14232192.
- ↑ Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP (2005). "MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks". Cell. 123 (7): 1213–26. doi:10.1016/j.cell.2005.09.038. PMID 16377563.
- ↑ Chapman JR, Jackson SP (2008). "Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage". EMBO Rep. 9 (8): 795–801. doi:10.1038/embor.2008.103. PMC 2442910. PMID 18583988.
- ↑ Luijsterburg MS, Acs K, Ackermann L, Wiegant WW, Bekker-Jensen S, Larsen DH, Khanna KK, van Attikum H, Mailand N, Dantuma NP (2012). "A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure". EMBO J. 31 (11): 2511–27. doi:10.1038/emboj.2012.104. PMC 3365417. PMID 22531782.
- ↑ Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H (2010). "The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage". J. Cell Biol. 190 (5): 741–9. doi:10.1083/jcb.201001048. PMC 2935570. PMID 20805320.
- ↑ Hanahan D, Weinberg RA (January 2000). "The hallmarks of cancer". Cell. 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9. PMID 10647931. S2CID 1478778.
- ↑ Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O (July 1998). "Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer". Nature. 394 (6689): 203–6. Bibcode:1998Natur.394..203V. doi:10.1038/28212. PMID 9671307. S2CID 6019090.
- ↑ Shain AH, Pollack JR (2013). "The spectrum of SWI/SNF mutations, ubiquitous in human cancers". PLOS ONE. 8 (1): e55119. Bibcode:2013PLoSO...855119S. doi:10.1371/journal.pone.0055119. PMC 3552954. PMID 23355908.
- 1 2 Hodges C, Kirkland JG, Crabtree GR (August 2016). "The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer". Cold Spring Harbor Perspectives in Medicine. 6 (8): a026930. doi:10.1101/cshperspect.a026930. PMC 4968166. PMID 27413115.
- ↑ Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, Sanchez-Cespedes M (May 2008). "Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines". Human Mutation. 29 (5): 617–22. doi:10.1002/humu.20730. PMID 18386774. S2CID 8596785.
- 1 2 3 Hodges HC, Stanton BZ, Cermakova K, Chang CY, Miller EL, Kirkland JG, Ku WL, Veverka V, Zhao K, Crabtree GR (January 2018). "Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers". Nature Structural & Molecular Biology. 25 (1): 61–72. doi:10.1038/s41594-017-0007-3. PMC 5909405. PMID 29323272.
- 1 2 Stanton BZ, Hodges C, Calarco JP, Braun SM, Ku WL, Kadoch C, Zhao K, Crabtree GR (February 2017). "Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin". Nature Genetics. 49 (2): 282–288. doi:10.1038/ng.3735. PMC 5373480. PMID 27941795.
- ↑ Baliñas-Gavira, Carlos; Rodríguez, María I.; Andrades, Alvaro; Cuadros, Marta; Álvarez-Pérez, Juan Carlos; Álvarez-Prado, Ángel F.; de Yébenes, Virginia G.; Sánchez-Hernández, Sabina; Fernández-Vigo, Elvira; Muñoz, Javier; Martín, Francisco; Ramiro, Almudena R.; Martínez-Climent, José A.; Medina, Pedro P. (October 2020). "Frequent mutations in the amino-terminal domain of BCL7A impair its tumor suppressor role in DLBCL". Leukemia. 34 (10): 2722–2735. doi:10.1038/s41375-020-0919-5. ISSN 1476-5551. PMID 32576963.
- ↑ Andrades, Alvaro; Peinado, Paola; Alvarez-Perez, Juan Carlos; Sanjuan-Hidalgo, Juan; García, Daniel J.; Arenas, Alberto M.; Matia-González, Ana M.; Medina, Pedro P. (2023-02-21). "SWI/SNF complexes in hematological malignancies: biological implications and therapeutic opportunities". Molecular Cancer. 22 (1): 39. doi:10.1186/s12943-023-01736-8. ISSN 1476-4598. PMC 9942420. PMID 36810086.
- ↑ Liquori, Alessandro; Ibañez, Mariam; Sargas, Claudia; Sanz, Miguel Ángel; Barragán, Eva; Cervera, José (2020-03-08). "Acute Promyelocytic Leukemia: A Constellation of Molecular Events around a Single PML-RARA Fusion Gene". Cancers. 12 (3): 624. doi:10.3390/cancers12030624. ISSN 2072-6694. PMC 7139833. PMID 32182684.
- ↑ Wolffe AP (May 2001). "Chromatin remodeling: why it is important in cancer". Oncogene. 20 (24): 2988–90. doi:10.1038/sj.onc.1204322. PMID 11420713.
- ↑ Tu, William B.; Helander, Sara; Pilstål, Robert; Hickman, K. Ashley; Lourenco, Corey; Jurisica, Igor; Raught, Brian; Wallner, Björn; Sunnerhagen, Maria; Penn, Linda Z. (May 2015). "Myc and its interactors take shape". Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 1849 (5): 469–483. doi:10.1016/j.bbagrm.2014.06.002. ISSN 0006-3002. PMID 24933113.
- ↑ Marks PA, Dokmanovic M (December 2005). "Histone deacetylase inhibitors: discovery and development as anticancer agents". Expert Opinion on Investigational Drugs. 14 (12): 1497–511. doi:10.1517/13543784.14.12.1497. PMID 16307490. S2CID 1235026.
- ↑ Richon VM, O'Brien JP (2002). "Histone deacetylase inhibitors: a new class of potential therapeutic agents for cancer treatment" (PDF). Clinical Cancer Research. 8 (3): 662–4. PMID 11895892.
- ↑ Richon VM, Sandhoff TW, Rifkind RA, Marks PA (August 2000). "Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation". Proceedings of the National Academy of Sciences of the United States of America. 97 (18): 10014–9. Bibcode:2000PNAS...9710014R. doi:10.1073/pnas.180316197. PMC 27656. PMID 10954755.
- ↑ Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K, Hamaguchi M, Hara Y, Kobayashi S, Iwase H (November 2005). "Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*". Breast Cancer Research and Treatment. 94 (1): 11–6. doi:10.1007/s10549-005-6001-1. PMID 16172792. S2CID 27550683.
- ↑ Munshi, Anupama; Tanaka, Toshimitsu; Hobbs, Marvette L.; Tucker, Susan L.; Richon, Victoria M.; Meyn, Raymond E. (August 2006). "Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci". Molecular Cancer Therapeutics. 5 (8): 1967–1974. doi:10.1158/1535-7163.MCT-06-0022. ISSN 1535-7163. PMID 16928817. S2CID 26874948.
- ↑ "Zolinza® (vorinostat) Capsules. Full Prescribing Information" (PDF). Food and Drug Administration (FDA). Archived from the original (PDF) on 20 January 2022. Retrieved 9 February 2023.
- ↑ VanderMolen, Karen M.; McCulloch, William; Pearce, Cedric J.; Oberlies, Nicholas H. (August 2011). "Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma". The Journal of Antibiotics. 64 (8): 525–531. doi:10.1038/ja.2011.35. ISSN 1881-1469. PMC 3163831. PMID 21587264.
- ↑ "ISTODAX® (romidepsin) for injection, for intravenous use. Full prescribing information" (PDF). Food and Drug Administration (FDA). Archived from the original (PDF) on 25 January 2022. Retrieved 9 February 2023.
- ↑ Dowden J, Hong W, Parry RV, Pike RA, Ward SG (April 2010). "Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases". Bioorganic & Medicinal Chemistry Letters. 20 (7): 2103–5. doi:10.1016/j.bmcl.2010.02.069. PMID 20219369.
- ↑ Wong, Lee H.; McGhie, James D.; Sim, Marcus; Anderson, Melissa A.; Ahn, Soyeon; Hannan, Ross D.; George, Amee J.; Morgan, Kylie A.; Mann, Jeffrey R.; Choo, K. H. Andy (March 2010). "ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells". Genome Research. 20 (3): 351–360. doi:10.1101/gr.101477.109. ISSN 1549-5469. PMC 2840985. PMID 20110566.
- ↑ Clapier CR, Cairns BR (2009). "The biology of chromatin remodeling complexes". Annual Review of Biochemistry. 78: 273–304. doi:10.1146/annurev.biochem.77.062706.153223. PMID 19355820.
- 1 2 Parry, Aled John; Narita, Masashi (2016). "Old cells, new tricks: chromatin structure in senescence". Mammalian Genome. 27 (7–8): 320–331. doi:10.1007/s00335-016-9628-9. ISSN 0938-8990. PMC 4935760. PMID 27021489.
- ↑ Hayflick, L.; Moorhead, P. S. (1961-12-01). "The serial cultivation of human diploid cell strains". Experimental Cell Research. 25 (3): 585–621. doi:10.1016/0014-4827(61)90192-6. ISSN 0014-4827. PMID 13905658.
- 1 2 3 Chandra, Tamir; Ewels, Philip Andrew; Schoenfelder, Stefan; Furlan-Magaril, Mayra; Wingett, Steven William; Kirschner, Kristina; Thuret, Jean-Yves; Andrews, Simon; Fraser, Peter; Reik, Wolf (2015-01-29). "Global Reorganization of the Nuclear Landscape in Senescent Cells". Cell Reports. 10 (4): 471–483. doi:10.1016/j.celrep.2014.12.055. ISSN 2211-1247. PMC 4542308. PMID 25640177.
- 1 2 3 4 5 6 Sun, Luyang; Yu, Ruofan; Dang, Weiwei (2018-04-16). "Chromatin Architectural Changes during Cellular Senescence and Aging". Genes. 9 (4): 211. doi:10.3390/genes9040211. ISSN 2073-4425. PMC 5924553. PMID 29659513.
- 1 2 3 4 Criscione, Steven W.; Teo, Yee Voan; Neretti, Nicola (2016). "The chromatin landscape of cellular senescence". Trends in Genetics. 32 (11): 751–761. doi:10.1016/j.tig.2016.09.005. ISSN 0168-9525. PMC 5235059. PMID 27692431.
- 1 2 3 4 Yang, Na; Sen, Payel (2018-11-03). "The senescent cell epigenome". Aging (Albany NY). 10 (11): 3590–3609. doi:10.18632/aging.101617. ISSN 1945-4589. PMC 6286853. PMID 30391936.
- 1 2 Basta, Jeannine; Rauchman, Michael (2015). "The Nucleosome Remodeling and Deacetylase (NuRD) Complex in Development and Disease". Translational Research. 165 (1): 36–47. doi:10.1016/j.trsl.2014.05.003. ISSN 1931-5244. PMC 4793962. PMID 24880148.
- 1 2 Li, Xueping; Ding, Dong; Yao, Jun; Zhou, Bin; Shen, Ting; Qi, Yun; Ni, Ting; Wei, Gang (2019-07-15). "Chromatin remodeling factor BAZ1A regulates cellular senescence in both cancer and normal cells". Life Sciences. 229: 225–232. doi:10.1016/j.lfs.2019.05.023. ISSN 1879-0631. PMID 31085244. S2CID 155090903.
- 1 2 López-Otín, Carlos; Blasco, Maria A.; Partridge, Linda; Serrano, Manuel; Kroemer, Guido (2013-06-06). "The Hallmarks of Aging". Cell. 153 (6): 1194–1217. doi:10.1016/j.cell.2013.05.039. ISSN 0092-8674. PMC 3836174. PMID 23746838.
- 1 2 Tasdemir, Nilgun; Banito, Ana; Roe, Jae-Seok; Alonso-Curbelo, Direna; Camiolo, Matthew; Tschaharganeh, Darjus F.; Huang, Chun-Hao; Aksoy, Ozlem; Bolden, Jessica E.; Chen, Chi-Chao; Fennell, Myles (2016). "BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance". Cancer Discovery. 6 (6): 612–629. doi:10.1158/2159-8290.CD-16-0217. ISSN 2159-8290. PMC 4893996. PMID 27099234.
- ↑ Wilson, V. L.; Jones, P. A. (1983-06-03). "DNA methylation decreases in aging but not in immortal cells". Science. 220 (4601): 1055–1057. Bibcode:1983Sci...220.1055W. doi:10.1126/science.6844925. ISSN 0036-8075. PMID 6844925.
- ↑ Kaeberlein, Matt; McVey, Mitch; Guarente, Leonard (1999-10-01). "The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms". Genes & Development. 13 (19): 2570–2580. doi:10.1101/gad.13.19.2570. ISSN 0890-9369. PMC 317077. PMID 10521401.
Further reading
- Chen T, Dent SY (February 2014). "Chromatin modifiers and remodellers: regulators of cellular differentiation". Nature Reviews Genetics. 15 (2): 93–106. doi:10.1038/nrg3607. PMC 3999985. PMID 24366184.
External links
- MBInfo - Chromatin
- MBInfo - DNA Packaging
- YouTube - Chromatin, Histones and Modifications
- YouTube - Epigenetics Overview
- Chromatin+remodeling at the U.S. National Library of Medicine Medical Subject Headings (MeSH)